

Osteoporosis is traditionally characterized as a systemic skeletal disorder defined by low bone mineral density and microarchitectural deterioration. However, viewing this pathology merely as a macro-level calcium deficiency or an inevitable consequence of aging oversimplifies the highly complex cellular disruptions driving it. At the molecular level, osteoporosis represents a homeostatic network failure within the Basic Multicellular Unit (BMU) the transient anatomical structure where bone resorption and bone formation are tightly coupled.
In the pathological marrow microenvironment, this coupling is severed. The rate of osteoclastic resorption outpaces osteoblastic bone formation, leading to a net loss of trabecular connectivity and cortical thickness. Devising advanced biological interventions requires moving past outdated concepts of physical cell replacement. Modern regenerative medicine focuses on the therapeutic deployment of Stem Cell Therapy Bangkok Thailand and extracellular vesicle (EV) arrays. Rather than engrafting to structurally replace tissue, these paracrine vectors operate as molecular master keys that reprogram the local cellular niche.
Pathological Signaling Disruptions vs Stem Cell Therapy Bangkok Thailand
To understand how targeted biological signaling alters the trajectory of bone loss, we must examine the specific molecular friction points within the bone marrow niche.
| Pathological State (Osteoporotic Niche) | Secretome-Mediated State (Regenerative Niche) |
| Hyper-activated RANKL signaling accelerating osteoclastogenesis. | High-affinity Osteoprotegerin (OPG) delivery acting as a competitive decoy receptor. |
| Lineage misdirection shifting progenitor cells from osteogenesis to adipogenesis. | Exosomal microRNAs driving epigenetic upregulation of the Wnt/β-catenin pathway. |
| Microvascular decay and loss of specialized H-type (CD31hiEmcnhi) capillaries. | Paracrine upregulation of VEGF and PDGF-BBrestoring angiogenic-osteogenic coupling. |
| Chronic Senescence-Associated Secretory Phenotype (SASP) maintaining a pro-inflammatory tone. | Targeted delivery of IL-10 and TGF-β resetting the neuroimmune microenvironment. |
Deep-Dive Mechanisms of Paracrine-Mediated Bone Restoration
1. Attenuation of the RANKL/OPG Axis & Osteoclastogenesis Arrest
The primary driver of accelerated bone resorption, particularly in estrogen-deficient and senescent states, is the upregulation of Receptor Activator of Nuclear Factor-B Ligand (RANKL). Secreted by osteocytes and hypertrophic T-cells under stress, RANKL binds to its cognate receptor RANK on the surface of myeloid progenitor cells. This binding triggers the recruitment of TRAF6, activating downstream cascades including NF-B and MAPK pathways, which ultimately translocate the master transcription factor NFATc1. This process rapidly drives the fusion, maturation, and survival of hyper-functional osteoclasts.
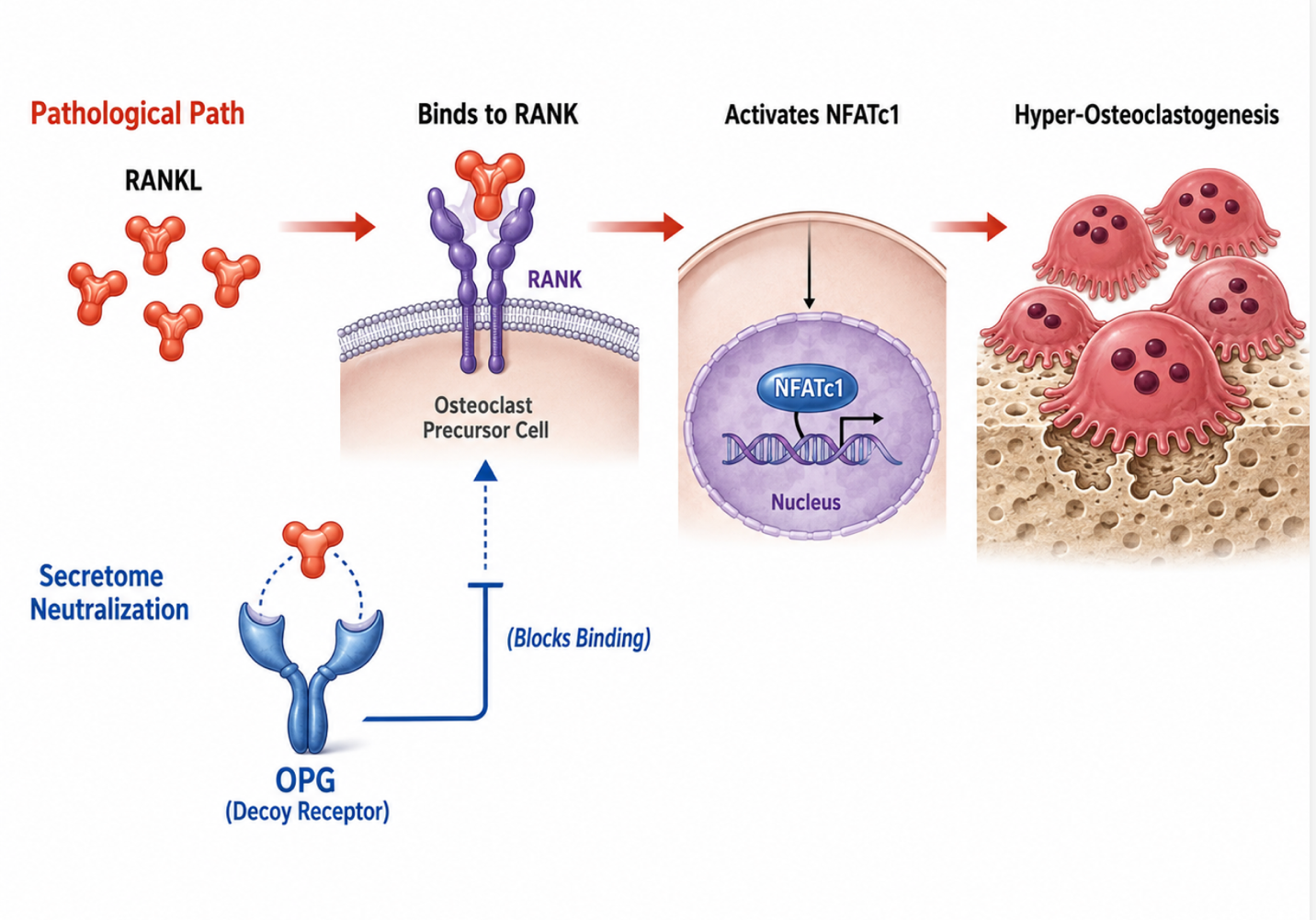
Figure 1: Secretome-Derived OPG as a Decoy Receptor Blocking RANKL–RANK Signaling and Hyper-Osteoclastogenesis
Secretome fractions derived from high-potency neonatal mesenchymal lineages interrupt this destructive cascade through the targeted delivery of Osteoprotegerin (OPG). OPG functions as a soluble, high-affinity decoy receptor that binds explicitly to RANKL before it can interface with native RANK receptors. By competitively neutralizing circulating RANKL, the secretome restores the physiological RANKL/OPG balance. This structural interception halts downstream NFATc1 translocation, effectively arresting osteoclast differentiation at the progenitor stage, decreasing the osteoclast life span, and stopping active trabecular excavation.
2. Epigenetic Lineage Reprogramming via Exosomal microRNAs
In the osteoporotic bone marrow niche, resident Stem Cell Therapy Bangkok Thailand experience a profound lineage shift known as the osteogenic-to-adipogenic switch. Age-related oxidative stress and altered cytokine profiles cause these endogenous progenitors to favor differentiation into marrow adipocytes rather than bone-forming osteoblasts. This misdirection results in a fatty infiltration of the bone marrow cavity, concurrent with a drastic drop in the recruitment of functional osteoblasts capable of synthesizing the type I collagen matrix.
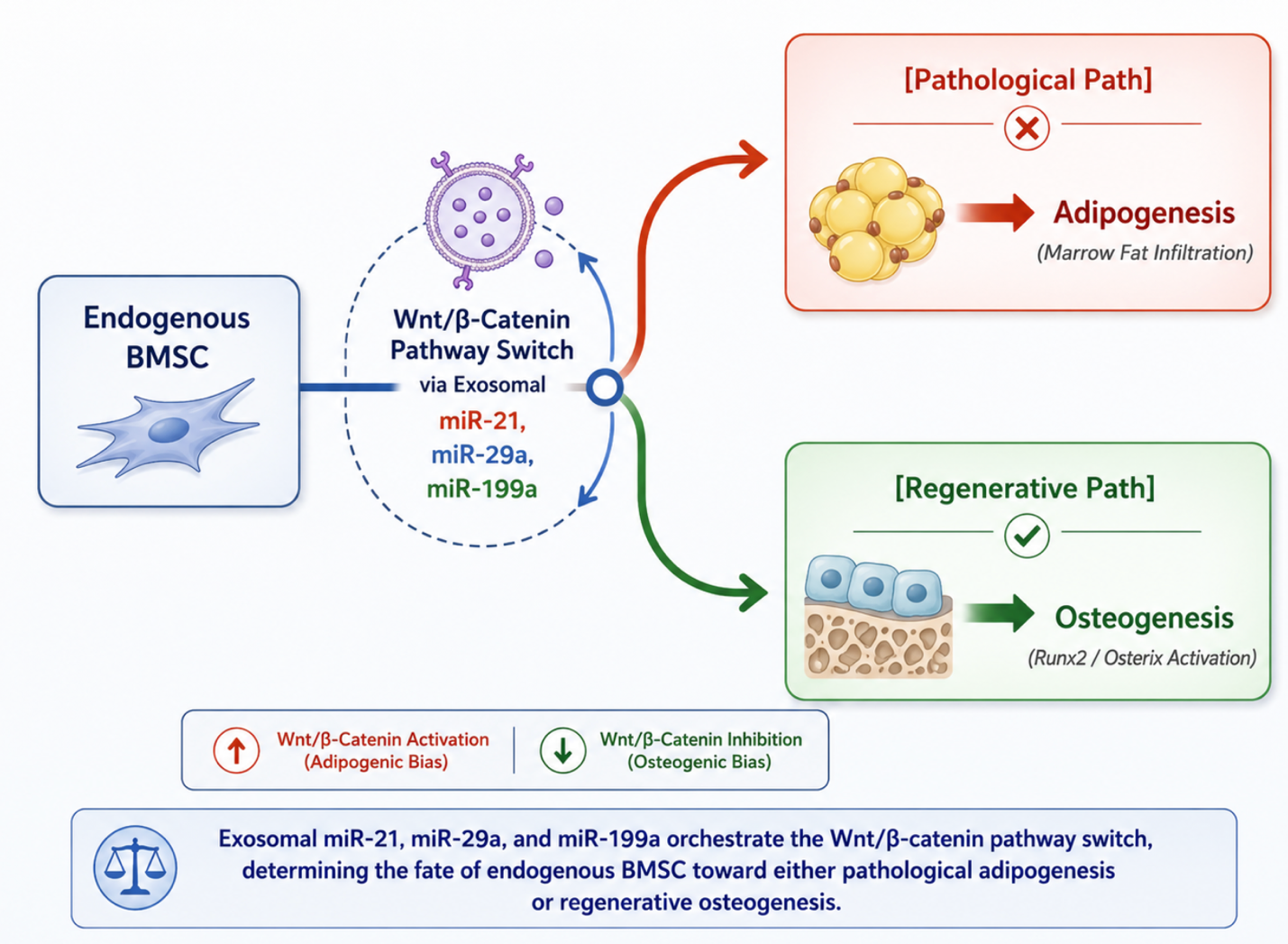
Figure 2: Exosomal miRNA-Mediated Wnt/β-Catenin Pathway Switching in Endogenous BMSC Fate Determination
Advanced cellular interventions counter this lineage drift through extracellular vesicle (EV) and exosome communication. These nano-scale vesicles pass through the lipid bilayers of exhausted host BMSCs to deliver a specialized cargo of regulatory microRNAs predominantly miR-21, miR-29a, and miR-199a. These microRNAs act as epigenetic modifiers that suppress endogenous Wnt inhibitors, such as Dickkopf-1 (DKK1) and Sclerostin (SOST). By clearing these inhibitory checkpoints, the exosomal payload forcefully reactivates the intracellular Wnt/-catenin signaling pathway. Accumulated -catenin translocates into the nucleus, directly upregulating the transcription of Runt-related transcription factor 2 (Runx2) and Osterix (Osx). This molecular switch shifts the progenitor pool away from fat production, guiding them into becoming active, mineralizing osteoblasts.
3. Restoration of Angiogenic-Osteogenic Coupling via H-Type Capillaries
Bone remodeling is highly dependent on spatial structural organization; it cannot occur without precise spatial coupling between vascular networks and Stem Cell Therapy Bangkok Thailand. In the osteoporotic skeleton, the microvascular architecture of the bone marrow undergoes severe degradation. Specifically, there is a systemic loss of a specialized capillary subtype known as H-type vessels, structurally defined by high co-expression of CD31 and Endomucin (). H-type capillaries are uniquely capable of generating a specific metabolic and molecular microenvironment that guides bone formation, as their perivascular cells secrete critical survival factors for young osteoblasts.
The Stem Cell Therapy Bangkok Thailand array directly targets this vascular decay by deploying potent pro-angiogenic factors, including Vascular Endothelial Growth Factor (VEGF) and Platelet-Derived Growth Factor-BB (PDGF-BB). PDGF-BB signaling induces the migration and proliferation of pericytes, while VEGF drives the directional sprouting of endothelial cells. Together, these paracrine signals induce the recrudescence of the vascular architecture within the trabecular junctions. These newly formed H-type capillaries serve as physical structural pathways and signaling hubs, importing essential nutrients, removing metabolic waste, and recruiting fresh osteoprogenitor cells directly to the remodeling surfaces to accelerate bone matrix mineralization.
4. Marrow Niche SASP Deactivation & Neuroimmune Rebalancing
Chronic skeletal degradation is increasingly understood to be driven by a state of persistent low-grade inflammation within the bone marrow cavity, a phenomenon termed “inflammaging.” Driven by systemic estrogen withdrawal and cellular aging, resident immune and stromal cells transition into a senescent state. Here, they exhibit a Senescence-Associated Secretory Phenotype (SASP), continuously secreting pro-inflammatory cytokines such as TNF-, IL-1$beta$, and IL-6. This toxic inflammatory tone directly degrades the bone extracellular matrix, upregulates RANKL expression on osteocytes, and induces apoptosis in surviving osteoblasts.
Advanced cell-free secretome signaling systematically counteracts this chronic inflammation by introducing highly concentrated anti-inflammatory cytokines, specifically Interleukin-10 (IL-10) and Transforming Growth Factor-beta (TGF-).
Mechanism of Action: IL-10 binds to its cell-surface receptor complex (IL-10R1/IL-10R2), initiating the JAK1/STAT3 signaling cascade. This intracellular pathway actively represses the transcription of pro-inflammatory genes within host macrophages and T-cells, effectively deactivating the local SASP loop.
By neutralizing the inflammatory pressure, the secretome restores neuroimmune equilibrium within the marrow niche. This shift establishes a stable environment that allows newly formed bone matrix to mature and cross-link without being broken down by chronic inflammatory stress.
The Investigational Paradigm: Navigating Clinical Boundaries
When exploring advanced biological care for systemic skeletal conditions, maintaining clinical clarity is vital. While the cellular mechanisms of secretome signaling, exosomal microRNA transfer, and angiogenic-osteogenic coupling are well-documented in preclinical models and peer-reviewed literature, they must be accurately framed within current clinical boundaries.
These advanced signaling applications are classified as supportive and investigational biological modalities. They are designed to optimize the molecular health of the bone marrow microenvironment and should not be promoted as definitive, standalone cures or guaranteed reversals of severe structural osteoporosis. Responsible clinical integration avoids sensationalist terminology like “immediate cell replacement.” Instead, it positions these therapies as sophisticated biological tools deployed alongside standard orthopedic management, metabolic optimization, and targeted physical rehabilitation to comprehensively protect skeletal architecture.