

Umbilical cord mesenchymal stromal cells (UC-MSCs) are one of the most widely used platforms in regenerative medicine due to their immunomodulatory capability, paracrine signaling potential, and comparative manufacturing advantage over select adult tissue derivatives. In 2026, though, the landscape is no longer shaped so much by broad therapeutic enthusiasm as by concerns about product consistency, potency, manufacturing control and regulatory credibility. Recent reviews reaffirm the biological rationale for MSC-based interventions yet highlight outstanding limitations of donor variability, culture conditions, passage-dependent changes leading to bioprocessing diversity, release testing and comparability across clinical trials. On December 18, 2024, the U.S. Food and Drug Administration (FDA) approved remestemcel-L-rknd (Ryoncil), which was the first FDA-approved mesenchymal stromal cell therapy but bone marrow-derived rather than umbilical cord-derived. Simultaneously, consensus efforts that have emerged in recent years have contributed to more standardized definitions and reporting guidelines for MSC-based therapeutics, which is part of a larger shift toward reproducible and transparent reporting. This review eloquently details the current state of UC-MSC knowledge, including consideration of mechanism, clinical development stage, manufacturing consistency and potency assessment standards, regulatory oversight practices governing use or delivery of these cells (allogenic vs. autologous), as well as a concurrent rise in cell-free MSC-derived products like ECMs and secretomes.
- Introduction
Hence, umbilical cord mesenchymal stromal cells are still the subject of intensive investigation due to their attractive biological characteristics together with practical translational advantages. MSCs are thought to affect their tissue environment by direct cell-cell interactions and the secretion of cytokines, growth factors, and extracellular vesicles thus regulating inflammation, angiogenesis and repair related signaling (Contemporary reviews on MSCs). But in 2026, the big question is no longer whether UC-MSCs are biologically interesting. It is whether they can be reproducibly manufactured and meaningfully characterized, and evaluated in trials-with enough rigor to allow dependable clinical use.
- Why UC-MSCs Continue to Attract Interest
UC-MSCs are attractive as perinatal tissues provide a relatively easy access source of allogeneic stromal cells with high proliferative potential and wide immunomodulatory significance. Recent reviews refer to UC-MSCs’ ability to shape an inflammatory microenvironment through soluble immunomodulatory mediators and exosome-based extracellular vesicles (EVEs), and also highlight that source-specific differences can affect phenotype as well as functional status. This is relevant, as the therapeutic identity of an UC-MSC product relies not only on tissue origin but also on isolation, expansion and release methods for the cells.
- Present Clinical Position: Promise, but Uneven Evidence
The current clinical status of UC-MSCs is most appropriately described as promising and heterogeneous. Wide-ranging MSC reviews in 20257 remain to address potential applications across a spectrum of graft-vs-host disease, inflammatory disorders, liver disease, neurologic conditions, wound repair and various other indications but have reiterated inconsistent efficacy across studies and persistent translational barriers to routine translation. This distinction is important even for a journal-style discussion, because biological plausibility and compelling early-phase signals are present, but there remains significant variation in strength of evidence between diseases, dosing strategies, routes of delivery, and product manufacturing approaches.
- A Regulatory Inflection Point for the MSC Field
A significant event in the field occurred on December 18, 2024, when remestemcel-L-rknd (Ryoncil) was approved by the FDA for steroid-refractory acute graft-versus-host disease in pediatric patients. FDA characterized Ryoncil as an allogeneic, bone marrow-derived MSC therapy, and specifically stated it was the first FDA-approved MSC therapy. While this product is not based on UC-MSCs, and while the approval itself has scope beyond that originally established by the concept of MSC therapy from a single tissue source, signifying a transition between a field driven largely by experimental positioning to one increasingly defined in terms of therapeutic products via established manufacturing quality paradigms complemented by regulatory-grade evidence.
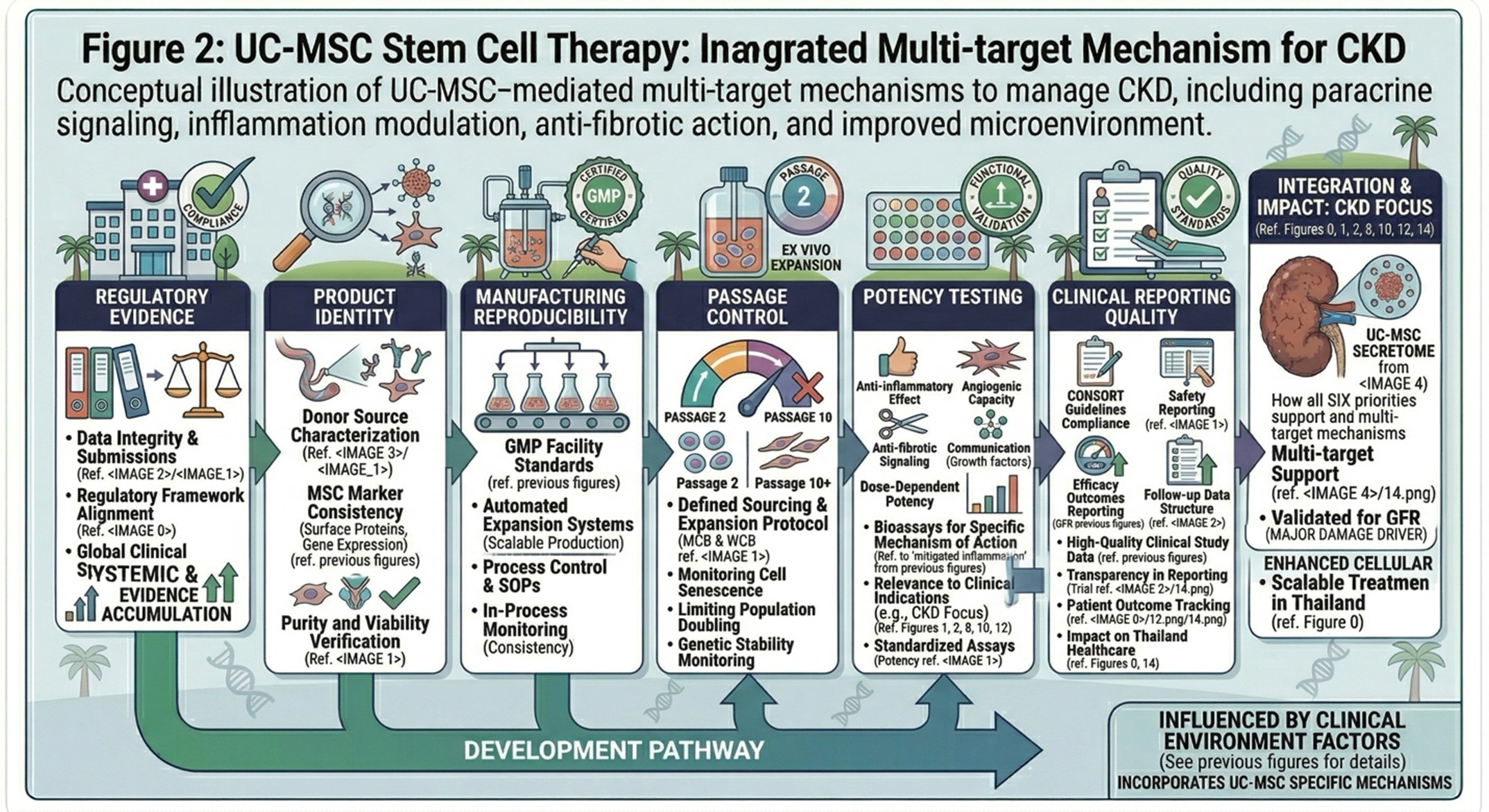
- Manufacturing Has Become a Central Scientific Question
It is no longer a background issue in 2026. Critically, conventional fetal bovine serum-based systems have fallen out of favor for translational MSC production due to concerns regarding reproducibility, safety, scalability and regulatory acceptance, and now reviews highlighting aspects of interventional products are biased toward describing serum-free,xeno-free and chemically defined culture systems. Design of experience large-scale expansion methods and processes is being seen more and more as part of product quality than just technical logistics. This evolution reflects the maturation of the field: the translational value of UC-MSCs is now as much dependent on controlled bioprocessing as it is on therapeutic concept.
- Passage Number, Heterogeneity, and Functional Drift
Passage-dependent change is one of the most practical issues today. A recent narrative review for UC-MSC passage number was the first to report that in vitro expansion has been shown to change its proliferation, senescence, immunophenotype, genomic stability, differentiation behavior and functional suitability. That same review mentions early-to-middle passages as the most sensible compromise between scalability and preserved therapeutic properties. This is a reminder that two products labeled “UC-MSCs” may differ from each other even before clinical variables have been considered.
- Potency Assays and the Push for Standardization
Potency is considered one of the top bottlenecks in MSC translation. An especially notable recent initiative in this regard was formed through consensus work in Cytotherapy that used a Delphi-based process to create and define a consensus definition of MSC types and to develop reporting guidance for use in MSC clinical studies with the specific intention of enhancing transparency, reproducibility, and comparability throughout the field. The translational message is unambiguous: viability and surface-marker identity are insufficient. If clinical findings are to be compared meaningfully across studies and (in the case of product) across manufacturing lots, products increasingly need functionally relevant assays and better-defined release frameworks.
- Parallel Development: Extracellular Vesicles and Secretome-Based Products
A related avenue of the field has been the development of MSC-derived extracellular vesicles (EVs) and secretome products. Knock-out reviews have reported significant interest in such cell-free approaches as they could potentially sustain some of the paracrine activity of MSCs while at the same time enhancing storage, dosing flexibility and product handling 20. Though, at the same time, these reviews highlight significant translational barriers such as inconsistent dose metrics, absence of validated potency assays and manufacturing complexity. EV and secretome development needs to be viewed as an extension of the standardization challenge facing the MSC field, rather than subconsciously an automatic fix therein.
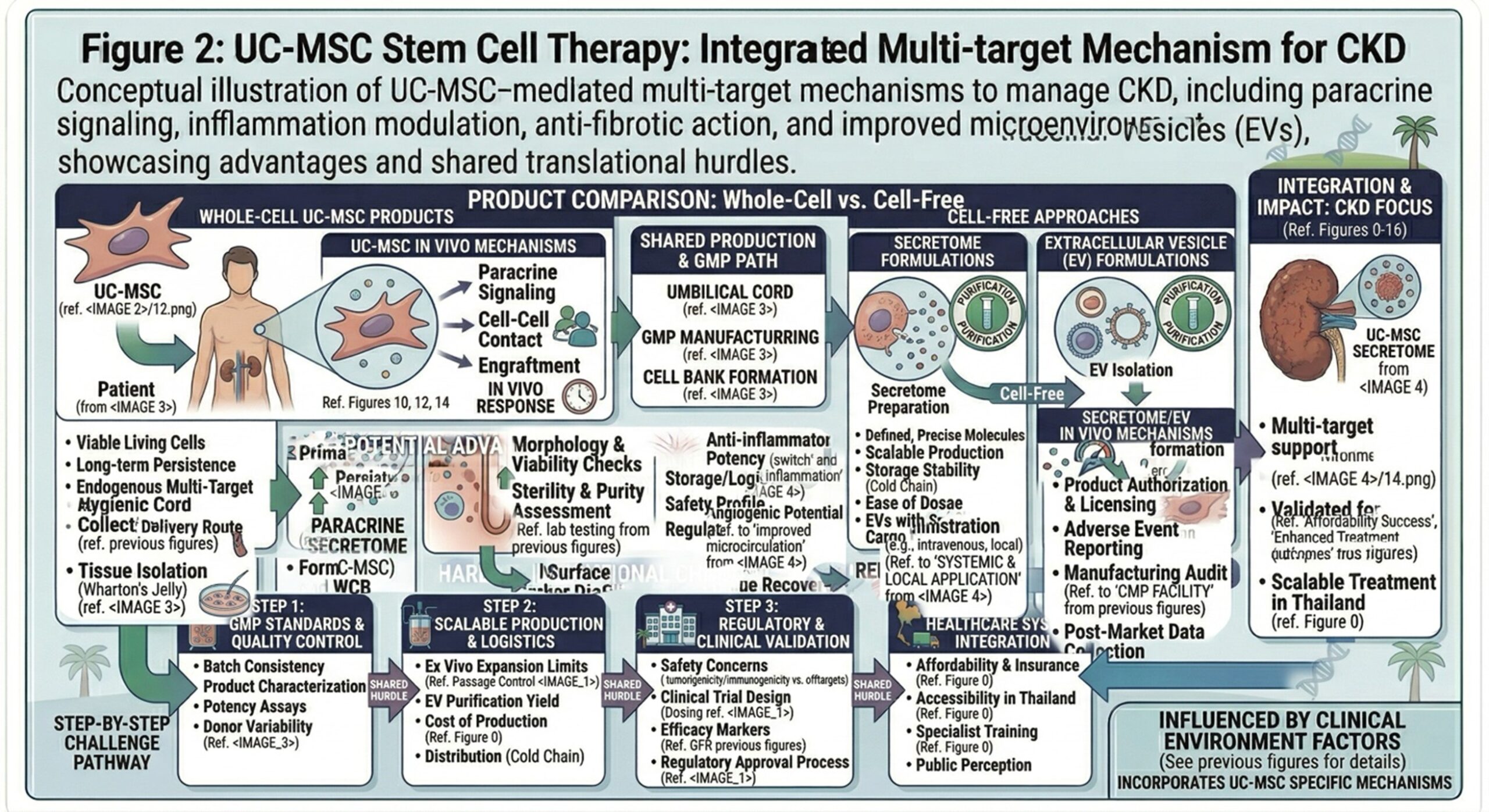
- Safety, Oversight, and the Difference Between Approved and Unapproved Products
The discussion around safety has also sharpened. On March 12, 2026, the FDA further warned the public that patients and consumers face risks from unapproved human tissue products marketed online for a multitude of diseases, as they have generally never been reviewed or proven to be adequate in quality, safety, purity or potency. At the same time, the ISSCR’s 2025 guidelines increasingly stress responsible clinicians wishing to translate their work into practice through rigorous oversight, and oppose any unproven stem cell intervention. Taken together, these documents or announcements reinforce the truism for UC-MSCs in 2026: The label “stem cell” does not bestow equal evidentiary or regulatory standing on products.
- Conclusion
The most accurate snapshot of UC-MSCs in 2026 is that the field has not been able to solve its core problems. It is that the field has gotten more rigorous about pinpointing them. This overview of UC-MSCs emphasizes their biological appeal and clinical utility in many lines of examination but also highlights that the future of this platform will be driven by reproducible manufacture, passage-aware process control, credible potency assays, transparent reporting as well as alignment with regulators. And in that regard, the current moment isn’t characterized by stronger claims but tougher standards.